ゲノミクス
ゲノミクス
ゲノミクスは、ゲノムの構造、機能、進化、マッピング、編集に焦点を当てた学際的な生物学の分野です。ゲノムは、そのすべての遺伝子を含む、生物のDNAの完全なセットです。 個々の遺伝子とその遺伝における役割の研究を指す遺伝学とは対照的に、ゲノミクスは、生物の遺伝子のすべて、それらの相互関係の集合的な特性評価と定量化を目的としています。と生物への影響。遺伝子は、酵素とメッセンジャー分子の助けを借りてタンパク質の生産を指示するかもしれません。次に、タンパク質は臓器や組織などの体の構造を構成し、化学反応を制御し、細胞間で信号を伝達します。ゲノミクスには、ハイスループットDNAシーケンスとバイオインフォマティクスを使用して、ゲノム全体の機能と構造を組み立てて分析することにより、ゲノムのシーケンスと分析も含まれます。ゲノミクスの進歩は、発見に基づく研究とシステム生物学に革命を引き起こし、脳などの最も複雑な生物学的システムの理解を促進しました。
この分野には、ゲノム内(ゲノム内)現象の研究も含まれます。エピスタシス(ある遺伝子が別の遺伝子に及ぼす影響)、多面性(1つの遺伝子が複数の形質に影響を与える)、ヘテロシス(ハイブリッド活力)、およびゲノム内の遺伝子座と対立遺伝子間のその他の相互作用など。
内容
歴史
系統学
ギリシャ語のΓΕΝ gen から、「遺伝子」(ガンマ、イプシロン、nu、イプシロン)は「なる、 create、creation、birth」、およびその後のバリアント:遺伝子学、創世記、遺伝学、遺伝子、ゲノム、遺伝子型、属など。ゲノムという単語(ドイツ語のゲノムから)、ハンスウィンクラーに起因する)は1926年には英語で使用されていましたが、ゲノムという用語は、ジャクソン研究所(メイン州バーハーバー)の遺伝子学者であるトムロデリックが開催された会議でビールを使って造語しました。私n1986年のヒトゲノムのマッピングに関するメリーランド州。
初期の配列決定の取り組み
ロザリンドフランクリンによるDNAのらせん構造の確認に続いて、ジェームズD.ワトソンとフランシスクリックによる1953年のDNAの構造と、1955年のフレッドサンガーによるインスリンのアミノ酸配列の発表により、核酸配列決定は初期の分子生物学者の主要な標的となりました。 1964年、Robert W. Holleyらは、これまでに決定された最初の核酸配列であるアラニン転移RNAのリボヌクレオチド配列を発表しました。この作業を拡張して、マーシャル・ニーレンバーグとフィリップ・レダーは、遺伝暗号の三重項の性質を明らかにし、実験で64個のコドンのうち54個の配列を決定することができました。 1972年、ゲント大学(ベルギー、ゲント)の分子生物学研究所のWalter Fiersと彼のチームは、遺伝子の配列を最初に決定しました。バクテリオファージMS2コートタンパク質の遺伝子です。 Fiersのグループは、MS2コートタンパク質の研究を拡大し、1976年と1978年にバクテリオファージMS2-RNA(ゲノムは3569塩基ペアで4つの遺伝子のみをコードする)とシミアンウイルス40の完全なヌクレオチド配列を決定しました。
開発されたDNAシーケンシング技術
インスリンのアミノ酸配列に関する独創的な研究に加えて、フレデリックサンガーと彼の同僚は、包括的な確立を可能にするDNAシーケンシング技術の開発において重要な役割を果たしました。ゲノムシーケンシングプロジェクト。 1975年、彼とAlan Coulsonは、放射性標識ヌクレオチドを含むDNAポリメラーゼを使用したシーケンス手順を発表しました。これは、彼がプラスとマイナスの手法と呼んでいます。これには、定義された3 '末端を持つ短いオリゴヌクレオチドを生成する2つの密接に関連した方法が含まれていました。これらは、ポリアクリルアミドゲルでの電気泳動(ポリアクリルアミドゲル電気泳動と呼ばれる)によって分画され、オートラジオグラフィーを使用して視覚化することができます。この手順では、一度に最大80ヌクレオチドをシーケンスでき、大きな改善でしたが、それでも非常に面倒でした。それにもかかわらず、1977年に彼のグループは一本鎖バクテリオファージφX174の5,386ヌクレオチドのほとんどを配列決定することができ、最初の完全に配列決定されたDNAベースのゲノムを完成させました。 Plus and Minus メソッドの改良により、チェーンターミネーション、つまりサンガー法(以下を参照)が生まれました。これは、DNAシーケンス、ゲノムマッピング、データストレージ、およびバイオインフォマティクス分析の手法の基礎を形成しました。次の四半世紀の研究で最も広く使用されています。同じ年に、ハーバード大学のWalterGilbertとAllanMaxamは、既知の塩基でのDNAの優先的な切断を含む、DNAシーケンシングのMaxam-Gilbert法(化学的方法としても知られる)を独自に開発しました。効率的な方法。核酸の配列決定における画期的な研究で、ギルバートとサンガーは1980年のノーベル化学賞の半分をポールバーグ(組換えDNA)と共有しました。
完全なゲノム
これらのテクノロジーの出現により、ゲノムシーケンシングプロジェクトの範囲と完了速度が急速に強化されました。真核生物のオルガネラの最初の完全なゲノム配列であるヒトミトコンドリア(16,568 bp、約16.6 kb)は、1981年に報告され、最初の葉緑体ゲノムは1986年に続きました。1992年、最初の真核生物の染色体、醸造用酵母の染色体III Saccharomyces cerevisiae (315 kb)の配列が決定されました。シーケンスされた最初の自由生活生物は、1995年のインフルエンザ菌(1.8 Mb)の生物でした。翌年、北米、ヨーロッパ、および日本の研究所の研究者のコンソーシアムが、真核生物の最初の完全なゲノム配列、 S。 cerevisiae (12.1 Mb)であり、それ以来、ゲノムは指数関数的に増加するペースでシーケンスされ続けています。 2011年10月の時点で、2,719のウイルス、1,115の古細菌と細菌、36の真核生物の完全な配列が利用可能であり、そのうちの約半分は真菌です。
ゲノムが完全に配列決定された微生物のほとんどは問題があります。 Haemophilusinfluenzae などの病原体は、微生物の多様性の幅と比較して、それらの系統発生的分布に顕著な偏りをもたらしました。他の配列決定された種のうち、それらがよく研究されたモデル生物であるか、または良いモデルになることが約束されたので、ほとんどが選ばれました。酵母( Saccharomyces cerevisiae )は長い間真核細胞の重要なモデル生物でしたが、ミバエキイロショウジョウバエは非常に重要なツールでした(特に初期の前分子において)遺伝学)。ワーム Caenorhabditis elegans は、多細胞生物によく使用される単純なモデルです。ゼブラフィッシュ Brachydanio rerio は、分子レベルでの多くの発達研究に使用されており、植物 Arabidopsisthaliana は顕花植物のモデル生物です。日本のフグ( Takifugu rubripes )と斑点を付けられた緑のフグ( Tetraodon nigroviridis )は、ほとんどの種に比べてノンコーディングDNAがほとんど含まれていない、小さくコンパクトなゲノムのために興味深いものです。 。哺乳類の犬( Canisfamiliaris )、ドブネズミ( Rattus norvegicus )、マウス( Mus musculus )、チンパンジー( Pantroglodytes) )はすべて医学研究における重要なモデル動物です。
ヒトゲノムの大まかなドラフトは2001年の初めにヒトゲノムプロジェクトによって完成され、多くのファンファーレを生み出しました。 2003年に完了したこのプロジェクトは、特定の1人のゲノム全体のシーケンスを行い、2007年までにこのシーケンスは「終了」と宣言されました(20,000塩基で1つ未満のエラー、すべての染色体が組み立てられました)。それ以来、2012年10月に1,092ゲノムの配列決定を発表した1000人ゲノムプロジェクトの支援もあり、他の多くの個人のゲノムが配列決定されました。このプロジェクトの完了は、劇的に多くの開発によって可能になりました。効率的なシーケンシング技術と、大規模な国際協力による重要なバイオインフォマティクスリソースのコミットメントが必要でした。人間のゲノムデータの継続的な分析は、人間社会に深刻な政治的および社会的影響を及ぼしています。
「オミクス」革命
英語の新造語オミクスは、非公式に研究分野を指します。ゲノミクス、プロテオミクス、メタボロミクスなど、 -omics で終わる生物学。関連する接尾辞-omeは、それぞれゲノム、プロテオーム、メタボロームなど、このような分野の研究対象に対応するために使用されます。分子生物学で使用される接尾辞 -ome は、ある種の全体を指します。同様に、オミクスは一般的に大規模で包括的な生物学的データセットの研究を指すようになりました。この用語の使用の増加により、一部の科学者(Jonathan Eisenなど)は売られ過ぎだと主張するようになりましたが、これは、のすべての構成要素の完全またはほぼ完全な品揃えの定量分析に対する方向性の変化を反映しています。システム。たとえば、共生の研究では、かつては単一の遺伝子産物の研究に限定されていた研究者が、複数の種類の生物学的分子の全補体を同時に比較できるようになりました。
ゲノム解析
生物が選択された後、ゲノムプロジェクトには、DNAの配列決定、元の染色体の表現を作成するためのその配列の組み立て、およびその表現の注釈と分析の3つのコンポーネントが含まれます。
シーケンス
歴史的に、シーケンシングは、研究を含むシーケンシングセンター、一元化された施設(年間数十テラベースをシーケンシングするJoint Genome Instituteなどの大規模な独立機関から地域の分子生物学コア施設まで)で行われていました。費用のかかる機器と技術サポートが必要な研究所。しかし、シーケンシング技術が向上し続けるにつれて、新世代の効果的な高速ターンアラウンドベンチトップシーケンサーが平均的な学術研究室の手の届くところに来ました。全体として、ゲノムシーケンシングアプローチは、ショットガンとハイスループット(または次世代)シーケンシングの2つの大きなカテゴリに分類されます。
ショットガンシーケンシングは、1000塩基対より長く、染色体全体を含むDNA配列の分析用に設計されたシーケンシング方法です。これは、ショットガンの急速に拡大する準ランダムな発砲パターンとの類推によって名付けられました。ゲル電気泳動シーケンシングはかなり短いシーケンス(100〜1000塩基対)にしか使用できないため、長いDNAシーケンスをランダムな小さなセグメントに分割し、シーケンスして読み取りを取得する必要があります。この断片化とシーケンシングを数回実行することにより、ターゲットDNAの複数のオーバーラップリードが取得されます。次に、コンピュータプログラムは、異なる読み取りの重複する端を使用して、それらを連続したシーケンスにアセンブルします。ショットガンシーケンスはランダムサンプリングプロセスであり、特定のヌクレオチドが再構築されたシーケンスで表されるようにするには、オーバーサンプリングが必要です。ゲノムがオーバーサンプリングされる平均読み取り数は、カバレッジと呼ばれます。
その歴史の多くにおいて、ショットガンシーケンスの基礎となる技術は、古典的なチェーンターミネーション法または「サンガー法」でした。これは、invitroでのDNA複製中にDNAポリメラーゼが鎖末端ジデオキシヌクレオチドを選択的に組み込むことに基づいています。最近、ショットガンシーケンシングは、特に大規模な自動ゲノム解析のために、ハイスループットシーケンシング法に取って代わられました。ただし、サンガー法は、主に小規模なプロジェクトや、特に長い連続したDNA配列読み取り(500ヌクレオチド以上)を取得するために広く使用されています。鎖終結法には、一本鎖DNAテンプレート、DNAプライマー、DNAポリメラーゼ、通常のデオキシヌクレオシド三リン酸(dNTP)、およびDNA鎖伸長を終結させる修飾ヌクレオチド(dideoxyNTP)が必要です。これらの鎖末端ヌクレオチドは、2つのヌクレオチド間のホスホジエステル結合の形成に必要な3'-OH基を欠いており、ddNTPが組み込まれるとDNAポリメラーゼがDNAの伸長を停止します。 ddNTPは、DNAシーケンサーで検出するために放射性または蛍光標識することができます。通常、これらのマシンは、1日最大48回の実行で1回のバッチ(実行)で最大96個のDNAサンプルをシーケンスできます。
低コストのシーケンスに対する高い需要により、ハイスループットシーケンスの開発が促進されています。シーケンシングプロセスを並列化し、一度に数千または数百万のシーケンスを生成するテクノロジー。ハイスループットシーケンシングは、標準的な色素ターミネーター法で可能なものを超えてDNAシーケンシングのコストを下げることを目的としています。超ハイスループットシーケンシングでは、500,000もの合成によるシーケンシング操作を並行して実行できます。
イルミナのダイシーケンシング法は、リバーシブルダイターミネーターに基づいており、1996年にジュネーブ生物医学研究所、パスカルメイヤーとローランファリネッリによる。この方法では、最初にDNA分子とプライマーをスライドに付着させ、ポリメラーゼで増幅して、最初は「DNAコロニー」と呼ばれる局所的なクローンコロニーを形成します。配列を決定するために、4種類の可逆的ターミネーター塩基(RT塩基)が追加され、組み込まれていないヌクレオチドが洗い流されます。パイロシーケンシングとは異なり、DNA鎖は一度に1ヌクレオチド延長され、画像取得は遅延した瞬間に実行できるため、単一のカメラから取得したシーケンシャル画像で非常に大きなDNAコロニーのアレイをキャプチャできます。酵素反応と画像キャプチャを分離することで、最適なスループットと理論的に無制限のシーケンス容量が可能になります。最適な構成では、機器の最終的なスループットはカメラのA / D変換率のみに依存します。カメラは蛍光標識されたヌクレオチドの画像を撮影し、次に末端の3 'ブロッカーとともに色素がDNAから化学的に除去され、次のサイクルが可能になります。
別のアプローチであるイオン半導体シーケンシングは、標準的なDNA複製化学に基づいています。この技術は、塩基が組み込まれるたびに水素イオンの放出を測定します。テンプレートDNAを含むマイクロウェルは単一のヌクレオチドで溢れています。ヌクレオチドがテンプレート鎖に相補的である場合、それは組み込まれ、水素イオンが放出されます。このリリースにより、ISFETイオンセンサーがトリガーされます。ホモポリマーがテンプレート配列に存在する場合、複数のヌクレオチドが単一のフラッドサイクルに組み込まれ、検出された電気信号は比例して高くなります。
アセンブリ
シーケンスアセンブリとは、整列を指します。元の配列を再構築するために、はるかに長いDNA配列のフラグメントをマージします。現在のDNAシーケンシングテクノロジーはゲノム全体を連続シーケンスとして読み取ることはできず、使用するテクノロジーに応じて20〜1000塩基の小さな断片を読み取るため、これが必要になります。 PacBioやOxfordNanoporeなどの第3世代シーケンシングテクノロジーは、通常、長さが10kb以上のシーケンシングリードを生成します。ただし、エラー率は約15%と高くなっています。通常、リードと呼ばれる短いフラグメントは、ショットガンシーケンシングゲノムDNAまたは遺伝子転写物(EST)から生じます。
アセンブリは、大きく2つのアプローチに分類できます。 de novo アセンブリ、過去に配列決定されたものと類似していないゲノム、およびアセンブリ中の参照として密接に関連する生物の既存のシーケンスを使用する比較アセンブリ。比較アセンブリと比較して、 de novo アセンブリは計算が難しく(NP困難)、ショートリードNGSテクノロジーにはあまり適していません。 de novo アセンブリパラダイムには、アセンブリの2つの主要な戦略、オイラーパス戦略とオーバーラップレイアウトコンセンサス(OLC)戦略があります。 OLC戦略は、最終的に、NP困難な問題であるオーバーラップグラフを介してハミルトンパスを作成しようとします。オイラーパス戦略は、deBruijnグラフを介してオイラーパスを見つけようとするため、計算がより扱いやすくなります。
完成したゲノムは、各レプリコンを表すあいまいさのない単一の連続したシーケンスを持つものとして定義されます。
注釈
DNA配列アセンブリだけでは、追加の分析なしではほとんど価値がありません。ゲノムアノテーションは、生物学的情報を配列に付加するプロセスであり、次の3つの主要なステップで構成されます。
- タンパク質をコードしないゲノムの部分を特定する
- 上の要素を特定するゲノム、遺伝子予測と呼ばれるプロセス、および
- 生物学的情報をこれらの要素に添付します。
自動注釈ツールは、これらの手順をインシリコ、人間の専門知識と潜在的な実験的検証を伴う手動注釈(別名キュレーション)とは対照的です。理想的には、これらのアプローチは同じアノテーションパイプライン内で共存し、相互に補完します(以下も参照)。
従来、アノテーションの基本レベルは、BLASTを使用して類似性を見つけ、相同体に基づいてゲノムにアノテーションを付けることです。 。最近では、注釈プラットフォームに追加情報が追加されています。追加情報により、手動アノテーターは、同じアノテーションが付けられた遺伝子間の不一致をデコンボリューションできます。一部のデータベースは、ゲノムコンテキスト情報、類似性スコア、実験データ、および他のリソースの統合を使用して、サブシステムアプローチを通じてゲノム注釈を提供します。他のデータベース(Ensemblなど)は、キュレーションされたデータソースと、自動化されたゲノムアノテーションパイプラインのさまざまなソフトウェアツールの両方に依存しています。 構造アノテーションは、ゲノム要素、主にORFとその局在、または遺伝子構造の識別で構成されます。 機能アノテーションは、生物学的情報をゲノム要素に添付することで構成されます。
シーケンスパイプラインとデータベース
関連する大量のデータの再現性と効率的な管理の必要性ゲノムプロジェクトでは、計算パイプラインがゲノミクスにおいて重要なアプリケーションを持っていることを意味します。
研究分野
機能ゲノミクス
機能ゲノミクスは、遺伝子(およびタンパク質)の機能と相互作用を説明するために、ゲノムプロジェクト(ゲノムシーケンスプロジェクトなど)によって生成された膨大な量のデータを利用しようとする分子生物学の分野です。機能ゲノミクスは、DNA配列や構造などのゲノム情報の静的な側面とは対照的に、遺伝子の転写、翻訳、タンパク質間相互作用などの動的な側面に焦点を当てています。機能ゲノミクスは、遺伝子、RNA転写物、およびタンパク質産物のレベルでDNAの機能に関する質問に答えようとします。機能ゲノミクス研究の重要な特徴は、これらの質問に対するゲノム全体のアプローチであり、一般に、従来の「遺伝子ごとの」アプローチではなく、ハイスループット手法が含まれます。
ゲノミクスの主要な分野はまださまざまな生物のゲノムの配列決定に関心がありますが、完全なゲノムの知識は、主にさまざまな条件での遺伝子発現のパターンに関係する機能ゲノミクスの分野の可能性を生み出しました。ここで最も重要なツールは、マイクロアレイとバイオインフォマティクスです。
構造ゲノミクス
構造ゲノミクスは、特定のゲノムによってコードされるすべてのタンパク質の3次元構造を記述しようとします。このゲノムベースのアプローチは、実験的アプローチとモデリングアプローチの組み合わせによる構造決定のハイスループット法を可能にします。構造ゲノミクスと従来の構造予測の主な違いは、構造ゲノミクスは、特定の1つのタンパク質に焦点を合わせるのではなく、ゲノムによってコードされるすべてのタンパク質の構造を決定しようとすることです。全ゲノム配列が利用可能であるため、実験的アプローチとモデリングアプローチの組み合わせにより、構造予測をより迅速に行うことができます。特に、多数のシーケンスされたゲノムと以前に解決されたタンパク質構造が利用可能であるため、科学者は以前に解決された構造のタンパク質構造をモデル化できます。ホモログ。構造ゲノミクスは、ゲノム配列を使用する実験方法や、既知の構造のタンパク質に対する配列または構造の相同性に基づく、または相同性のないタンパク質の化学的および物理的原理に基づくモデリングベースのアプローチを含む、構造決定への多数のアプローチを取ることを含みます。既知の構造。従来の構造生物学とは対照的に、構造ゲノミクスの取り組みによるタンパク質構造の決定は、タンパク質の機能に関して何かが知られる前に行われることがよくあります(常にではありません)。これにより、構造バイオインフォマティクスに新たな課題が生じます。つまり、3D構造からタンパク質の機能を決定することです。
エピゲノミクス
エピゲノミクスは、細胞の遺伝物質に対するエピジェネティックな修飾の完全なセットの研究です。 、エピゲノムとして知られています。エピジェネティックな修飾は、細胞のDNAまたはヒストンに対する可逆的な修飾であり、DNA配列を変更することなく、遺伝子発現に影響を与えます(Russell 2010 p.475)。最も特徴的なエピジェネティック修飾の2つは、DNAメチル化とヒストン修飾です。エピジェネティックな修飾は、遺伝子の発現と調節に重要な役割を果たし、分化/発達や腫瘍形成などの多くの細胞プロセスに関与しています。グローバルレベルでのエピジェネティクスの研究は、ゲノムハイスループットアッセイの適応によってごく最近可能になりました。
メタゲノミクス
メタゲノミクスはメタゲノム、環境サンプルから直接回収された遺伝物質。広い分野は、環境ゲノミクス、エコゲノミクス、またはコミュニティゲノミクスと呼ばれることもあります。従来の微生物学および微生物ゲノム配列決定は、培養されたクローン培養に依存していますが、初期の環境遺伝子配列決定は、特定の遺伝子(多くの場合16S rRNA遺伝子)をクローン化して、天然サンプルの多様性のプロファイルを生成します。そのような研究は、微生物の生物多様性の大部分が栽培ベースの方法によって見落とされていたことを明らかにしました。最近の研究では、「ショットガン」サンガーシーケンスまたは超並列パイロシーケンスを使用して、サンプリングされたコミュニティのすべてのメンバーからすべての遺伝子のほぼ偏りのないサンプルを取得しています。メタゲノミクスは、これまで隠されていた微視的生命の多様性を明らかにする力があるため、生物界全体の理解に革命をもたらす可能性のある微生物界を表示するための強力なレンズを提供します。
モデルシステム
バクテリオファージは、細菌の遺伝学および分子生物学において重要な役割を果たしてきました。歴史的に、それらは遺伝子構造と遺伝子調節を定義するために使用されていました。また、配列決定された最初のゲノムはバクテリオファージでした。しかし、バクテリオファージの研究は、明らかに細菌ゲノミクスによって支配されているゲノミクス革命を導きませんでした。ごく最近になって、バクテリオファージゲノムの研究が目立つようになり、それによって研究者はファージの進化の根底にあるメカニズムを理解できるようになりました。バクテリオファージのゲノム配列は、単離されたバクテリオファージの直接配列決定によって取得できますが、微生物ゲノムの一部として導出することもできます。細菌ゲノムの分析は、かなりの量の微生物DNAがプロファージ配列とプロファージ様要素からなることを示しています。これらの配列の詳細なデータベースマイニングは、細菌ゲノムの形成におけるプロファージの役割への洞察を提供します。全体として、この方法は多くの既知のバクテリオファージグループを検証し、細菌ゲノムからプロファージの関係を予測するための便利なツールになります。
現在、全ゲノム配列が利用できる24のシアノバクテリアがあります。これらのシアノバクテリアのうち15は、海洋環境に由来します。これらは、6つの Prochlorococcus 株、7つの海洋 Synechococcus 株、 Trichodesmium erythraeum IMS101、および Crocosphaera watsonii WH8501です。いくつかの研究は、これらの配列を使用して、海洋シアノバクテリアの重要な生態学的および生理学的特性を推測する方法を示しています。ただし、現在進行中のゲノムプロジェクトは他にもたくさんあります。その中には、さらに Prochlorococcus と海洋の Synechococcus 分離株、 Acaryochloris と Prochloronがあります。 、N2固定糸状シアノバクテリア Nodularia spumigena 、 Lyngbya aestuarii 、 Lyngbya majuscula 、および海洋シアノバセリアに感染するバクテリオファージ。したがって、ゲノム情報の増大する本体は、比較アプローチを適用することによって、グローバルな問題に対処するためのより一般的な方法で利用することもできます。この分野における進歩のいくつかの新しくエキサイティングな例は、調節RNAの遺伝子の同定、光合成の進化的起源への洞察、または分析されたゲノムへの遺伝子水平伝播の寄与の推定です。
ゲノミクスの応用
ゲノミクスは、医学、バイオテクノロジー、人類学、その他の社会科学を含む多くの分野で応用を提供してきました。
ゲノム医学
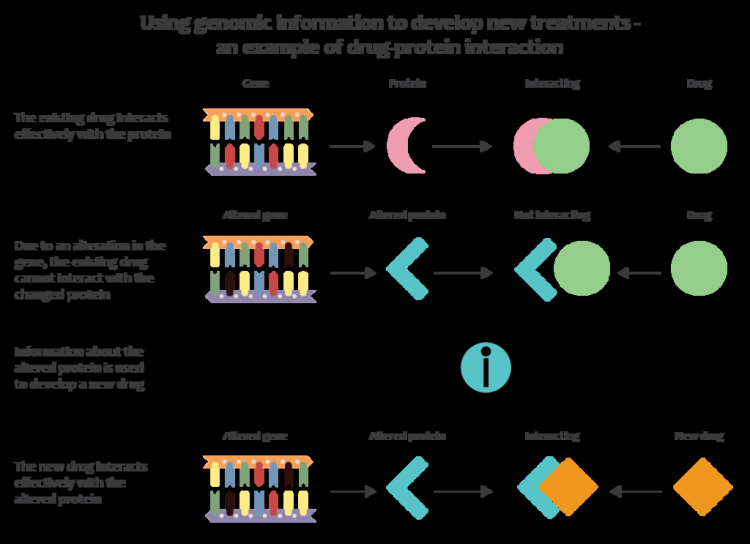
次世代ゲノミクステクノロジーにより、臨床医や生物医学研究者は、大規模な研究集団で収集されるゲノムデータの量を大幅に増やすことができます。病気の研究で多くの種類のデータをゲノムデータと統合する新しいインフォマティクスアプローチと組み合わせると、研究者は薬物反応と病気の遺伝的基盤をよりよく理解することができます。ゲノムを医学に適用するための初期の取り組みには、ヒトゲノムの医学的解釈のための最初のツールを開発したEuanAshleyが率いるスタンフォードチームによるものが含まれていました。たとえば、 All of Us の研究プログラムは、100万人の参加者からゲノム配列データを収集し、精密医学研究プラットフォームの重要なコンポーネントになることを目的としています。
合成生物学と生物工学
ゲノム知識の成長により、合成生物学のますます洗練されたアプリケーションが可能になりました。 2010年、J。CraigVenter Instituteの研究者は、 Mycoplasma genitalium のゲノムに由来する部分的に合成された細菌の種 Mycoplasma labatorium の作成を発表しました。
保存ゲノミクス
保存学者は、ゲノム配列決定によって収集された情報を使用して、集団の遺伝的多様性や個人が劣性遺伝子に対してヘテロ接合であるかどうかなど、種の保存に重要な遺伝的要因をより適切に評価できます。遺伝性遺伝性疾患。ゲノムデータを使用して進化過程の影響を評価し、特定の個体群全体の変動のパターンを検出することで、保護活動家は、標準的な遺伝的アプローチで対処されていない変数ほど多くの変数を不明のままにすることなく、特定の種を支援する計画を立てることができます。
ケネディ上院議員が脳腫瘍の手術を受ける
6月2日月曜日(HealthDay News)—エドワード・M・ケネディ上院議員は先月診断された悪性脳腫瘍の手術を受ける予定でした。 76歳の上院議員はノースカ …

ケラチンとは何ですか? このタンパク質が髪を強く輝かせておくのにどのように役立つか
ケラチンはあなたの体が自然に生成する構造タンパク質であり、髪の毛のすべての基礎です。爪や肌にも存在します。あなたの髪の房では、それはアルファケラチンと呼ばれ、そ …
